




Matthias Rath y Linus Pauling
Journal of Orthomolecular Medicine 1991, 6:125-134
"Mi querido Kepler, ¿qué opina de los grandes filósofos a quienes he ofrecido miles de veces mostrar mis estudios, pero quienes, con la perezosa obstinación de una serpiente que acaba de darse un atracón, nunca han consentido mirar los planetas ni la luna ni a través del telescopio? Verdaderamente, igual que las serpientes cierran los oídos, así los hombres cierran sus ojos a la luz de la verdad." Galileo Galilei en una carta a Johannes Kepler, ca. 1630
El siguiente artículo fue remitido por Linus Pauling al "Proceedings of the National Academy of Sciences" el 23 de abril de 1991 y aceptado para publicación el 11 de junio. Bajo circunstancias cuestionables, esta decisión fue revocada más tarde por el editor. Somos conscientes de que esta retirada no fue la decisión de un individuo. Se produjo en interés de quienes, personal o económicamente, dependen del actual dogma de las enfermedades cardiovasculares humanas. Confiamos en que los historiadores científicos hagan el juicio apropiado sobre este interesante desarrollo.
Estamos en deuda con el Journal of Orthomolecular Medicine por la publicación de este artículo sin retraso y sabemos que esta decisión no redundará en desventaja para esta revista. Por encima de todo, estamos convencidos de que la publicación de este artículo es en interés de millones de pacientes e incluso de todos los seres humanos.
Resumen
La enfermedad cardiovascular humana (ECV) es consecuencia de la acumulación de la lipoproteína(a), Lp(a), más que de las lipoproteínas de baja densidad (LDL), en la pared vascular. En general, no se debe a los niveles plasmáticos de LDL, sino al nivel de Lp(a), que se forma en el hígado a partir de las LDL y la apo(a) en cantidades que vienen determinadas en gran medida por la velocidad de síntesis de la apo(a). Esta velocidad aumenta cuando las concentraciones de ascorbato son bajas. La ECV humana es fundamentalmente una enfermedad degenerativa causada por un déficit de ascorbato. Este déficit es consecuencia de la incapacidad de los seres humanos de sintetizar ascorbato endógeno, combinada con la ingestión insuficiente de ascorbato en la dieta. La deficiencia se agrava por defectos genéticos, como el defecto del receptor de las LDL, y por los factores de riesgo exógenos de ECV que inducen a un agotamiento añadido de ascorbato. El déficit de ascorbato se traduce en cambios morfológicos de la pared vascular. Con objeto de evitar las consecuencias fatales derivadas de un agotamiento extremo de ascorbato, como las hemorragias en el escorbuto, el déficit de ascorbato aumenta de manera simultánea la concentración plasmática de los factores de riesgo hemostáticos y vasoconstrictores, entre ellos la Lp(a) y el fibrinógeno. El déficit crónico de ascorbato lleva a la acumulación extracelular de Lp(a) y de fibrinógeno/fibrina, los signos patognomónicos de la lesión arteriosclerótica.
El deterioro subyacente de la pared del vaso se pone de manifiesto fundamentalmente en zonas de condiciones hemodinámicas alteradas, lo que induce a el infarto de miocardio y a el accidente cerebrovascular como manifestaciones predominantes de la ECV humana.
Por tanto, para los pacientes con enfermedad coronaria o cerebrovascular, el factor de riesgo principal es la inestabilidad de la pared del vaso debida al déficit de ascorbato, antes que los constituyentes plasmáticos. Por el contrario, los factores de riesgo del plasma desencadenan la manifestación de la enfermedad vascular periférica (EVP). En esta enfermedad, los constituyentes plasmáticos, como los radicales libres de oxígeno del humo del tabaco o las lipoproteínas ricas en triglicéridos oxidativamente modificados, ejercen un efecto nocivo sobre la pared vascular en la periferia, por lo que se desarrolla la EVP. El agotamiento del ascorbato del tejido vascular es también una condición previa para la manifestación de la EVP. La ECV humana es multifactorial. El déficit de ascorbato, sin embargo, es el denominador común de esta enfermedad. La elaborada teoría sobre la patogenia y el tratamiento de la enfermedad cardiovascular humana presentada en este artículo representa la solución al rompecabezas de esta enfermedad y debe conducir a mejorar la salud humana.
Abreviaturas
Lp(a), lipoproteína(a); apo(a), apoproteína(a); apoB, apoproteína B; LDL, lipoproteínas de baja densidad; VLDL, lipoproteínas de muy baja densidad; IDL, lipoproteínas de densidad intermedia; HDL, lipoproteínas de alta densidad; ECV, enfermedad cardiovascular; EVP, enfermedad vascular periférica.
Introducción
Hemos formulado recientemente la hipótesis de que la lipoproteína(a), Lp(a), es un sustituto del ascorbato, la vitamina C (1). Este concepto reveló el papel fisiológico de la Lp(a), así como nuevos enfoques terapéuticos. Sobre la base de trabajos anteriores y de pruebas clínicas y experimentales añadidas, presentamos ahora una teoría detallada sobre la ECV humana. La causa principal de la ECV humana es un déficit de vitamina ascorbato que lleva al depósito de Lp(a) y fibrinógeno/fibrina en la pared arterial. Aclaramos la interacción entre el ascorbato y la Lp(a) y presentamos un mecanismo patógeno que difiere de los conceptos existentes (2,3,4) en que es capaz de explicar las características exclusivas de la arteriosclerosis humana. Presentamos también las consideraciones profilácticas y terapéuticas que abren nuevas vías a la prevención y el tratamiento de la ECV.
El papel fundamental de la Lp(a) en la ECV humana
La Lp(a) fue descubierta en 1963 por Kare Berg (5). Es muy parecida a las LDL; su principal diferencia radica en la unión de una glucoproteína, la apo(a), mediante un puente disulfuro, a la apoproteína de las LDL, la apoB, lo que proporciona un área de superficie mayor a la esfera de la lipoproteína. La secuencia de cDNA de la apo(a) muestra una notable homología con la del plasminógeno (6), con repeticiones múltiples de la región kringle (en espiral) 4, un kringle 5 y un dominio de proteasa. Debido a esta homología, la apo(a) se ha denominado el eslabón perdido entre la arterogenesis y la trombogénesis (7).
Las pruebas de que la Lp(a), y no las LDL, es la lipoproteína responsable fundamentalmente de la arteriosclerosis fueron publicadas por el equipo de uno de nosotros en la Universidad de Hamburgo (8,9,10). En los estudios más exhaustivos publicados todavía hasta la fecha sobre el papel de la Lp(a) en la pared vascular humana, se encontró que la Lp(a), no las LDL, se acumula de manera selectiva en la pared vascular de los pacientes con ECV. Además, la acumulación extracelular de Lp(a) mostraba una estrecha correlación con el desarrollo de las placas de arteroma. Lo que es aún más importante, en varios centenares de secciones histológicas transversales procedentes de la arteria coronaria y la aorta humanas, la inmunotinción para la apoB sin tinción concomitante para la apo(a) era un acontecimiento raro, lo que indica que el depósito de LDL sola en la pared vascular se produce rara vez (9). El depósito de Lp(a) en la pared vascular se determinó mediante análisis inmunomorfométrico, porque los métodos de extracción sobrevaloran el papel de las LDL: una fracción importante de la Lp(a) se encuentra disociada en la pared vascular en apo(a) y la partícula semejante a las LDL, sobre todo en condiciones postmortem (8). Los primeros investigadores anteriores no consiguieron evidentemente distinguir entre las LDL y la Lp(a), de modo que la iniciación de las lesiones arterioscleróticas se atribuyó de manera incorrecta a las LDL.
Esta conclusión se confirmó recientemente en un estudio sobre los factores plasmáticos de riesgo en los pacientes con defectos heredados del receptor de las LDL (11). En los pacientes con hipercolesterolemia familiar, la incidencia de ECV se determinó de manera significativa por la concentración plasmática de Lp(a), sin que hubiera relación entre, por un lado, el colesterol total y el colesterol LDL en plasma y, por otro, las manifestaciones clínicas de la ECV. Ahora hay pruebas experimentales y clínicas sólidas de que la Lp(a) es un factor de riesgo más importante que el colesterol total o el colesterol LDL para la coronariopatía (12), el accidente cerebrovascular (13) y la reestenosis de los injertos venosos después de cirugía de bypass coronario (14). Por consiguiente, concluimos que la Lp(a) es la lipoproteína fundamentalmente responsable de la iniciación de la ECV humana. El papel de las LDL se caracteriza mejor como un factor de riesgo agravante de la ECV en pacientes con niveles plasmáticos simultáneamente elevados de Lp(a).
La conexión ascorbato-Lp(a)
Observamos que la Lp(a) se ha detectado fundamentalmente en el plasma del ser humano, de otros primates y de algunas pocas otras especies que han perdido la capacidad de sintetizar ascorbato y, por consiguiente, tienen bajos niveles de este último, en comparación con los animales con producción endógena de ascorbato. No descartamos, sin embargo, que en otras especies se encuentren también pequeñas cantidades de Lp(a). La pérdida de la capacidad de síntesis de ascorbato es consecuencia de una mutación genética en el gen que codifica para la enzima L-gulono-???lactona oxidasa; esta mutación se produjo hace unos 40 millones de años en un antepasado de los primates. Posteriormente la Lp(a) se convirtió en un importante constituyente plasmático en los primates y el ser humano. Nosotros proponemos que la Lp(a) es un sustituto del ascorbato. Viceversa, el ascorbato es un sustituto de la Lp(a), ya que en la mayoría de las especies la Lp(a) es sustituida por el ascorbato sin desventaja alguna. Anteriormente se suponía que la Lp(a) era fundamentalmente una partícula patógena y que las concentraciones plasmáticas de Lp(a) venían determinadas fundamentalmente por factores genéticos. Nuestra publicación de la conexión Lp(a)-ascorbato (1) marcó un punto de inflexión en el rumbo experimental y sugirió numerosas investigaciones. Posteriormente se demostró que el ascorbato, el agente reductor más potente presente normalmente en el organismo, y también agentes reductores sintéticos, como la N-acetil-cisteína (15), reducen los niveles plasmáticos de Lp(a). En un ensayo clínico realizado en paciente con ECV, un aumento de la ingestión de ascorbato redujo el nivel plasmático de Lp(a) (observaciones no publicadas).
Además, propusimos que la Lp(a) fortalece la pared vascular, en particular cuando hay déficit de ascorbato. A bajas concentraciones de ascorbato, se deteriora la síntesis de colágeno y de elastina, y el depósito de Lp(a) contribuye a controlar la inestabilidad resultante de la pared del vaso y a contener la progresión de la enfermedad. La apo(a), una macromolécula, compensaría este deterioro y su demostrada unión a los glucosaminoglucanos y a otros compuestos de la matriz extracelular sería beneficiosa. Además, se ha demostrado que la apo(a) se une con gran afinidad a la prolina y la hidroxiprolina y es probable que se una al colágeno y la elastina, macromoléculas ricas en esos residuos de aminoácidos. El aumento de la ingestión de ascorbato elimina la necesidad de que la Lp(a) fortalezca los vasos sanguíneos y, por tanto, el ascorbato puede sustituir a la Lp(a). Recientemente nosotros hemos sido capaces de confirmar que el ascorbato puede sustituir a la Lp(a) en el lugar del proceso mórbido. En ese estudio piloto, utilizamos la cobaya hipoascorbémica, un animal, como el ser humano, incapaz de sintetizar ascorbato, pero capaz de sintetizar apo(a). Cuando se les alimentó con pequeñas cantidades de ascorbato en la dieta, correspondientes aproximadamente a la ingestión humana habitual, esos animales desarrollaron rápidamente placas de ateroma y depósitos de Lp(a) en la pared vascular. Ingestiones mayores de ascorbato inhibían el depósito de Lp(a) en la pared arterial e impedía el desarrollo de aterosclerosis (16).
El ascorbato y la regulación de la Lp(a) plasmática
Los niveles plasmáticos de Lp(a) varían de unas personas a otras en hasta 1000 veces. Esta considerable variación es resultado en gran medida de los factores genéticos que determinan la síntesis de apo(a), pero también de los que influyen en la apoB y los lípidos. Tal vez se deba a que la modificación de los genes que controlan la síntesis de apo(a) al nivel óptimo no ha resultado todavía completamente eficaz, de modo que, en algunos individuos, esta síntesis ha sobrepasado la marca, predisponiéndoles a una ECV. Además de por factores genéticos, las concentraciones plasmáticas de Lp(a) están reguladas también por factores alimenticios, uno de ellos es la niacina, de la que se ha demostrado que reduce los niveles plasmáticos de Lp(a) (17). Otro factor alimenticio es el ascorbato. Hemos obtenido los resultados preliminares de que el ascorbato reduce in vitro la síntesis de apo(a) en células de hepatoma humano. El ascorbato también puede disminuir la reunión de la partícula Lp(a) al reducir la formación de puentes disulfuro entre la apo(a) y la apoB en el hígado.
El déficit de ascorbato, el perfil de riesgo de ECV y la Lp(a)
El agotamiento del ascorbato es el denominador metabólico común de los factores de riesgo endógenos y exógenos para la ECV. Muchos defectos genéticos están asociados con el déficit de ascorbato. Como consecuencia de un defecto genético, disminuyen las constantes de velocidad de ciertas reacciones metabólicas controladas por enzimas. Estas constantes de velocidad pueden verse incrementadas hacia valores normales aumentando las concentraciones de ciertos cofactores (18). En un intento por normalizar esas constantes de velocidad reducidas, se agotan el ascorbato y otros cofactores esenciales para las reacciones metabólicas. El ascorbato, una potente molécula reductora e hidroxilante, es destruido en esas reacciones. Por consiguiente, en el esfuerzo por controlar el daño causado por el defecto genético, se reduce el nivel de ascorbato, exacerbando los efectos nocivos generales de su déficit.
Uno de los defectos genéticos para los cuales están bien caracterizadas las etapas que inducen un agotamiento de ascorbato es el defecto del receptor de las LDL. En todas las expresiones de los receptores de las LDL (19), la inhibición de la 3-hidroxi-3-metilglutaril coenzima A reductasa en la síntesis del colesterol (20), la protección de las LDL contra la modificación oxidativa (21) y la estimulación de la 7 ?-hidroxilasa en el catabolismo del colesterol a ácidos biliares (22), interviene el ascorbato. Nosotros sugerimos que es el déficit de ascorbato la causa real de la ECV prematura asociada con esta enfermedad heredada, exacerbada por el defecto genético. En este contexto, es interesante el reciente estudio realizado por Seed y cols. en pacientes con hipercolesterolemia familiar (11). En dicho estudio, no se encontró correlación con los niveles elevados de LDL ni con el defecto genético subyacente del receptor de las LDL. Por tanto, el defecto genético que induce un déficit de ascorbato, combinado con la disposición genética a niveles elevados de Lp(a) aumentó de manera significativa el riesgo de ECV prematura.
Igual que los defectos genéticos, los factores de riesgo exógenos de ECV producen agotamiento de ascorbato. Las correlaciones observadas entre una dieta rica en grasas y el tabaquismo, por un lado, y la ECV por otro pueden explicarse también como consecuencia de un déficit de ascorbato inducido, causado por la destrucción del ascorbato en el catabolismo de los lípidos y el esfuerzo por detoxificar las sustancias del humo del tabaco. Sin un complemento alimenticio suficiente de ascorbato, los factores de riesgo de ECV, tanto exógenos como endógenos, agravan el déficit de ascorbato y aceleran el desarrollo de ECV.
El déficit de ascorbato y la pared vascular
La anascorbemia, el agotamiento total de ascorbato observado en el escorbuto, induce la pérdida completa de la integridad y la estabilidad de la pared vascular, así como la extravasación de la sangre al área perivascular. La hipoascorbemia produce la forma precoz de este deterioro. El endotelio vascular se ve directamente afectado por el déficit de ascorbato. Los rasgos que lo caracterizan son cambios en la morfología celular y la presencia de grandes huecos intercelulares. Estos cambios producen la pérdida de la función del endotelio como barrera entre la sangre y la pared vascular, un aumento de la permeabilidad y, consecuentemente, un aumento de la infiltración de los constituyentes sanguíneos en la pared vascular. La matriz extracelular de la pared también se ve afectada. El colágeno y la elastina, las principales macromoléculas de esta matriz, se forman a partir de sus precursores, el procolágeno y la proelastina, por hidroxilación de los residuos prolil y lisil. El déficit de ascorbato induce una hidroxilación incompleta y, por tanto, debilita la matriz extracelular. Se sabe que las alteraciones del endotelio y el aflojamiento del tejido conjuntivo son rasgos característicos de las placas de ateroma.
El déficit de ascorbato y las contramedidas metabólicas
Para limitar las consecuencias del déficit prolongado de ascorbato, se desarrollaron contramedidas metabólicas bajo una fuerte presión evolutiva. El efecto más perjudicial del déficit de ascorbato es la pérdida de sangre. Por tanto, para evitar la extravasación de sangre, el déficit de ascorbato desencadena una serie de reacciones metabólicas con el objetivo principal de inducir vasoconstricción y hemostasia. Por consiguiente, no sorprende que el déficit de ascorbato induzca prácticamente todos los factores de riesgo que predisponen a la aterogénesis y a la trombogénesis, la mayoría de ellos con importancia clínica inmediata. En la primera línea de defensa contra el riesgo de hemorragia perivascular, el aumento de los niveles de tromboxano y la reducción de los niveles de prostaciclina (23) y prostaglandina E producen vasoconstricción y hemostasia. Hemos demostrado que el déficit prolongado de ascorbato incrementa los niveles plasmáticos de fibrinógeno y Lp(a) y, en esta situación, las propiedades antifibrinolíticas de la Lp(a) (24) se vuelven beneficiosas. Somos conscientes de que no hay una relación de correspondencia exacta entre el ascorbato y la Lp(a). La Lp(a) constituye una etapa bastante tardía de la secuencia de reactantes de fase aguda o de factores de riesgo inducidos por el déficit de ascorbato. Debido a su depósito en la pared vascular, sin embargo, la Lp(a) es particularmente perjudicial. Las implicaciones terapéuticas son evidentes: el suplemento con ascorbato aumenta los niveles de prostaciclina y, posiblemente, de EDRF, el factor relajante derivado del endotelio. Este potente factor vasodilatador es idéntico al óxido nítrico, y el ascorbato puede preservar la forma activa del EDRF inhibiendo su oxidación a dióxido de nitrógeno. A la vez, el ascorbato reduce los niveles de tromboxano, fibrinógeno y Lp(a) y, por consiguiente, contribuye a una mejora fundamental del perfil de riesgo en cardiología clínica.
El papel de la Lp(a) y del fibinógeno en la pared vascular
En los estudios de Hamburgo, la Lp(a) se encontró depositada fundamentalmente con fibrinógeno/fibrina (10). Además, se ha demostrado que la Lp(a) se une al fibrinógeno/fibrina inmovilizados (25) y se han publicado pruebas de unión directa de la Lp(a) con el fibrinógeno/fibrina en la pared vascular (26). Todas esas observaciones pueden explicarse ahora. En el déficit de ascorbato, resulta evidente la necesidad de un aumento de la concentración plasmática de Lp(a) y fibrinógeno, de la unión de la Lp(a) al fibrinógeno/fibrina en la pared vascular y de su retención selectiva. Las propiedades hemostáticas de la Lp(a) y del fibrinógeno son necesarias para contrarrestar las consecuencias deletéreas del déficit de ascorbato. La Lp(a) actúa también en la contención de las enfermedades y la reparación de los tejidos. La degradación tisular inducida por radicales libres y por plasmina son vías de progresión de la enfermedad bien establecidas. La Lp(a) puede actuar como un inhibidor para las dos vías. Hemos sugerido que la apo(a), debido a sus muchos grupos disulfuro que pueden ser reducidos a tioles por el ascorbato, puede funcionar por sí misma como un antioxidante (1). Además, ahora sugerimos que, debido a su homología con la plasmina, la Lp(a) inhibe también la degradación tisular inducida por la plasmina. El contenido lipídico de la partícula de Lp(a) proporciona simultáneamente el substrato para la reparación celular. Con objeto de ejercer sus funciones fisiológicas, la Lp(a) es depositada como una partícula lipoproteica intacta y puede aislarse de la pared vascular (8). La acumulación extracelular de Lp(a) en la pared vascular es un mecanismo patógeno independiente de la ECV humana, que no coincide con las teorías según las cuales la captación y la degradación de lipoproteínas por las células emigrantes es un requisito previo para la aterogénesis.
Una teoría sobre la enfermedad cardiovascular humana

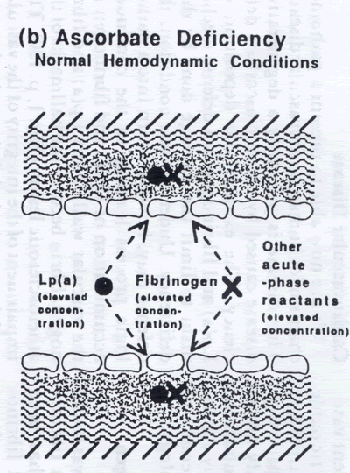
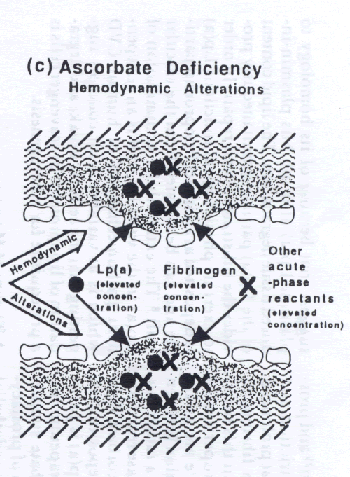
Es improbable que la Lp(a) ejerza fundamentalmente su aterogenicidad uniéndose al receptor del plasminógeno de las células endoteliales (27). Estos receptores están presentes por todo el sistema vascular, de modo que este mecanismo patógeno aumentaría la incidencia de enfermedades vasculares periféricas y de trombos venosos, que no estarían asociados necesariamente con niveles plasmáticos elevados de Lp(a).
Formas periféricas de arteriosclerosis
Ahora podemos explicar otro fenómeno asociado con la ECV humana, la diferencia principal entre los mecanismos patógenos que producen, por un lado, la arteriosclerosis en los puntos de predisposición y, por otro lado, la enfermedad vascular periférica (EVP). El infarto de miocardio y el accidente cerebrovascular son, con gran diferencia, las manifestaciones más frecuentes de la ECV. El desarrollo localizado de las placas de ateroma en esos pacientes sólo puede explicarse si la inestabilidad de la pared vascular es el principal factor de riesgo. Las concentraciones elevadas de los factores de riesgo plasmáticos, por ejemplo, el colesterol o las LDL, no pueden explicar el fenómeno de manifestación localizada de la ECV. Sin embargo, pueden desempeñar un papel agravante en el desarrollo de la ECV en el individuo.
En el desarrollo de la EVP, sin embargo, estos factores de riesgo plasmáticos desempeñan un papel mucho más destacado, ejerciendo un efecto nocivo directo o indirecto sobre la pared vascular. Por consiguiente, esto induce a la arteriosclerosis en la periferia vascular, donde el contacto entre los constituyentes plasmáticos nocivos y el endotelio es prolongado. Debido a su mayor sensibilidad a la peroxidación, las lipoproteínas ricas en triglicéridos serían esos posibles factores provocadores, induciendo a la lesión vascular en la periferia. Esta teoría explica la forma periférica de la ECV asociada con la hiperlipidemia de tipo III, un trastorno metabólico en el cual se acumulan lipoproteínas ricas en triglicéridos en el plasma en forma de VLDL y de IDL. Esas afecciones están caracterizadas también por otro mecanismo patógeno de depósito de lípidos en la pared vascular. Además del depósito extracelular de Lp(a) descrito antes, la captación celular de lipoproteínas oxidativamente modificadas por las células emigrantes desempeña un papel más destacado. Esto puede explicar también porqué las células espumosas se encuentran con mucha más frecuencia en la pared vascular de los pacientes con estos trastornos metabólicos. Un mecanismo patógeno similar interviene en la EVP asociada con el tabaquismo. Los radicales libres de oxígeno del humo del tabaco dañan el endotelio directamente o a través de modificación oxidativa de las lipoproteínas. Hay que destacar que el ascorbato, el antioxidante más potente normalmente presente en el organismo humano, también es un potente inhibidor de esos mecanismos patógenos. En general, los trastornos metabólicos hereditarios que se traducen en un aumento de la concentración de los constituyentes plasmáticos nocivos están asociados con frecuencia con la EVP, por ejemplo, en la homocistinuria. De particular interés es la patogenia de la EVP en la diabetes mellitus. Las moléculas de glucosa y de ascorbato comparten semejanzas estructurales y compiten por el mismo sistema de transporte para la captación celular. Niveles elevados de glucosa inhiben competitivamente la captación tisular óptima de ascorbato, produciendo también un agotamiento crónico de este último de la pared vascular y el deterioro de la pared. Por consiguiente, la administración de suplementos alimenticios de ascorbato debe producir un control eficaz de la angiopatía diabética.
De los diferentes mecanismos patogénicos implicados, el déficit de ascorbato es un denominador común de la ECV humana.
Consideraciones profilácticas y terapéuticas
La teoría presentada en este artículo sugiere inmediatamente los tratamientos profilácticos y terapéuticos eficaces para la mayoría de los individuos en situación de riesgo de ECV y de los pacientes con ECV.
Profilaxis
Se ha demostrado que el ascorbato, un potente agente reductor e hidroxilante, permite alcanzar de una manera eficaz los objetivos profilácticos: reducción del nivel plasmático de LP(a), prevención del depósito de Lp(a) en la pared vascular (16), reducción de los niveles elevados de LDL (28), aumento de los niveles de HDL (29), protección contra la lesión oxidante eliminando los radicales libres de oxígeno y regenerando el tocoferol, y evitando la modificación oxidativa de las lipoproteínas (30) y, sobre todo, preservando la integridad de la pared vascular e impidiendo la formación de las placas de arteroma (16). Además, el ascorbato actúa sobre todos estos objetivos al mismo tiempo. Es difícil que ningún producto farmacéutico supere al ascorbato, una sustancia que ha sido desarrollada y perfeccionada por la naturaleza a lo largo de miles de millones de años. La arteriosclerosis prematura es esencialmente desconocida en la mayoría de los animales, mientras que millones de seres humanos, con déficit crónico de ascorbato, mueren de arteriosclerosis y enfermedades relacionadas cada año.
Terapéutica
El ascorbato puede no sólo evitar la formación de lesiones arterioscleróticas, sino también reducir las placas existentes. Está bien establecido que el ascorbato aumenta los niveles de HDL, promoviendo con ello el transporte inverso del colesterol por captación de los lípidos intracelulares y extracelulares de la pared vascular. Basándonos en nuestro hallazgo de que el desarrollo de la placa se produce en paralelo con el depósito extracelular de Lp(a), es evidente que uno de los focos principales a los que debe dirigirse el desarrollo terapéutico es la liberación de la Lp(a), o su componente lipídico, de la pared arterial. El ascorbato puede intervenir de dos formas: disociando la apo(a) del componente semejante a las LDL de la Lp(a), intensificando así el flujo de salida de las lipoproteínas de la pared vascular y convirtiendo los residuos lisil de esta pared en residuos hidroxilisil, para reducir con ello la afinidad de unión a los componentes de la pared vascular por medio del grupo hapténico lisil. La eficacia de la liberación de la Lp(a) de sus enlaces al fibrinógeno/fibrina en la pared vascular puede verse considerablemente intensificada por la administración también de pequeñas dosis profilácticas o dosis terapéuticas mayores de uno o más de los inhibidores que compiten por los grupos hapténicos lisil (lisina, ácido 6-aminohexanoico, ácido p-aminometilciclohexano carboxílico y otros). Para los pacientes con ECV avanzada, podrían prescribirse cantidades terapéuticas de los inhibidores, junto con ascorbato, como coadyuvantes de un tratamiento convencional apropiado, una vez probado su efecto terapéutico.
Cabría sostener que esas sustancias, que suelen utilizarse como antifibrinolíticos, inducirían a complicaciones de la coagulación. Dichas sustancias son, sin embargo, inhibidores de la proteasa e inhiben la activación de la fibrinólisis, así como de la coagulación (31). Esas sustancias se han utilizado en estudios a largo plazo para diferentes indicaciones sin efectos secundarios comprometedores. Sin embargo, no hemos encontrado ninguna recomendación previa del uso de esas sustancias en el tratamiento farmacológico de la enfermedad cardiovascular. La combinación de esos inhibidores con el ascorbato puede considerarse ideal, ya que el ascorbato reduce la necesidad de depósito ulterior de la Lp(a) en la pared vascular y los inhibidores intensificarían la liberación de la Lp(a) ya depositada. Además, se sabe que el ascorbato tiene propiedades anticoagulantes (32) y profibrinolíticas.
Conclusión
El concepto presentado aquí ofrece por primera vez una explicación concluyente de las exclusivas características de la ECV humana. Puede responder a las preguntas para las cuales la hipótesis actualmente en vigor sobre el desarrollo de la ECV no ha proporcionado una explicación (12,3). El déficit de ascorbato es una condición previa, así como un denominador común, de la ECV. Con raras excepciones, la ECV es una enfermedad degenerativa. Su factor de riesgo principal es la inestabilidad de la pared vascular, antes que ningún constituyente plasmático, y su mecanismo patogénico fundamental es el despisto de Lp(a) y de fibrinógeno /fibrina. Ahora podemos explicar porqué la tendencia descendente más pronunciada en la mortalidad por ECV de todos los países industrializados se produjo en Estados Unidos, el país con el mayor consumo de vitamina C. Además, ahora entendemos porqué estos dos procesos se desarrallaron paralelamente. Según el concepto científico presentado en esta publicación, ahora es posible alcanzar un éxito similar también en otros países.
Los mecanismos patógenos descritos y las conclusiones terapéuticas presentadas aquí son la solución al rompecabezas de la ECV humana. Hemos comentado con detalle las siguientes cuestiones: la causa de la enfermedad actual más importante por déficit de ascorbato, el resultado de un defecto genético en combinación con una ingestión inadecuada de suplemento de ascorbato; la regulación de los niveles plasmáticos de Lp(a) por el ascorbato y las razones por las cuales la Lp(a) y el ascorbato se encuentran alterantivamente en la mayoría de las especies animales; la identificación del déficit de ascorbato como un denominador común de los factores de riesgo endógenos y exógenos de ECV; las condiciones bajo las cuales un mecanismo de defensa fisiológico diseñado por la naturaleza para limitar los efectos deletéreos del déficit de ascorbato puede convertirse en un proceso patológico; el depósito extracelular de Lp(a) y fibrinógeno/fibrina como el principal mecanismo de arteriogenesis humana; los detalles de una teoría exhaustiva sobre la enfermedad cardiovascular humana; y la diferencia entre la arteriosclerosis en los lugares de depósito y la enfermedad vascular periférica. Por último, presentamos las recomendaciones profilácticas y terapéuticas basadas en esos descubrimientos, que pueden llevar a un importante avance en la prevención y el tratamiento de la ECV humana.
La importancia terapéutica de nuestro descubrimiento no se limita a las ECV; la Lp(a) y el ascorbato están implicados en el cáncer, las enfermedades inflamatorias y otras enfermedades, e incluso el proceso del envejecimiento. El depósito de Lp(a) en las proximidades de la enfermedad puede concebirse como una mecanismo de defensa para contener la progresión de la enfermedad, en particular a concentraciones bajas de ascorbato. La conexión Lp(a)-ascorbato es un principio regulador de la naturaleza que afecta directamente a la salud humana. La abolición del déficit de ascorbato puede mejorar profundamente la salud humana e incrementar la esperanza de vida de los seres humanos.
Agradecimientos
Agradecemos a Roger Barth la supervisión del cuidado de los animales; a Dugui Jiang y Su-Cheng Yu su ayuda técnica; a Constance Tsao sus útiles comentarios; a Rosemary Babcock y James Liu su ayuda bibliográfica, a Jolanta Walichiewicz por su ayuda gráfica; y a Martha Best y Dorothy Munro su ayuda en secretaría con el manuscrito.
Bibliografía:
1. Rath M, Pauling L. (1990): Proc. Natl. Acad. Sci. USA, 87, 6204-6207.
2.Brown MS, Goldstein JL. (1984): Scientific American, 251, 58-66.
3.Ross R. (1986): N. Engl. J. Med. 314, 488-500.
4.Steinberg D, Parthasarathy S, Carew TE, Khoo JC, Witztum JL. (1989): N. Engl. J. Med. 320, 915-924.
5.Berg K. (1963): Acta Pathol. 59, 369-382.
6.McLean JW, Tomlinson JE, Kuang WJ, Eaton DL, Chen EY, Fless GM, Scanu AM, Lawn RM. (1987): Nature 300, 132-137.
7.Brown MS, Goldstein JL. (1987): Nature (London), 330, 113-114.
8.Rath M, Niendorf A, Reblin T, Dietel M, Kregger HJ, Beisiegel U (1989): Arteriosclerosis 9, 579-592.
9.Niendorf A, Rath M, Wolf K, Peters S, Arps H, Beisiegel U, Dietel M. (1990): Virchows Archiv. A. Pathol. Anat. 417, 105-111.
10.Beisiegel U, Niendorf A, Wolf K, Reblin T, Rath M. (1990): European Heart J. 11, Suppl. E., 174-183.
11.Seed BM, Hoppichler F, Reaveley D, McCarthy S, Thompson GR, Boerwinkle E, Utermann G. (1990): N. Engl. J. Med. 322, 1494-1499.
12.Dahlen GH, Guyton JR, Attar M, Farmer JA, Kautz JA, Gotto AM Jr. (1986): Circulation 74, 758-765.
13.Zenker G, Koltringer P, Bone G, Kiederkorn K, Pfeiffer K, Jurgens G. (1986): Stroke 17, 942-945.
14.Hoff HF, Gaubatz JW. (1982): Atherosclerosis 42, 273-297.
15.Gavish D, Breslow JL. (1991): Lancet 337, 203-204.
16.Rath M, Pauling L. (1990): Proc. Natl. Acad. Sci. USA 87, 9388-9390.
17.Carlson LA, Hamsten A, Asplund A. (1989): J. Intern. Med. 226, 271-276.
18.Pauling L. (1968): Science 160, 265-271.
19.Aulinskas TH, Van der Westerhuyzen DR, Coetzee GA. (1983): Atherosclerosis 47, 159-171.
20.Harwood HJ Jr., Greene YJ, Stacpoole PW. (1986): J. Biol. Chem. 261, 7127-7135.
21.Frei B, England L, Ames BN. (1989): Proc. Natl. Acad. Sci. USA 86, 6377-6381.
22.Ginter E. (1973): Science 179, 702-704.
23. Beetens J, Coene MC, Verheyen A, Zonnekyn L, Herman AG. (1986): Prostaglandins 32, 335-352.
24.Loscalzo J, Weinfeld M, Fless GM, Scanu AM. (1990): Arteriosclerosis 10, 240-245.
25.Harpel PC, Gordon BR, Parker TS. (1989): Proc. Natl. Acad. Sci USA 86, 3847-3851.
26.Smith EB, Cochran S. (1990): Atherosclerosis 84, 173-181.
27.Miles LA, Fless GM, Levin EG, Scanu AM, Plow EF. (1989): Nature 339, 301-303.
28.Ginter E. (1979): Wld. Rev. Nutr. Diet. 33, 104-141.
29.Bates CJ, Mandal AR, Cole TJ. (1977): Lancet 3, 611.
30.Jialal I, Vega GL, Grundy SM. (1990): Atherosclerosis 82, 185-191.
31.Aoki N, Naito K, Yoshida N. (1978): Blood 1, 1-12.
32.Bordia A, Verma, SD. (1985): Clin. Cardiol. 8, 552-554.
33.Paterson JC. (1941): Can. Med. Assoc. J. 44, 114-120.
34.Willis GC, Light AW, Gow WQS. (1954): Canad. Med. Ass. J. 71, 562-568.
vía
